固態(tài)電解質(zhì)
固態(tài)電解質(zhì)是通過一種原位熔化反應(yīng),在電解質(zhì)顆粒表面生成共價(jià)鍵配位,來解決固態(tài)電池的氧化穩(wěn)定性差和枝晶的問題。這種配位通過共價(jià)鍵合陰離子上的局部高濃度電子,從熱力學(xué)角度關(guān)閉了陰離子氧化分解過程中的電子交換,并從動(dòng)力學(xué)角度阻止了電解質(zhì)顆粒表面的電子滲流;這種現(xiàn)象導(dǎo)致了一個(gè)前所未有的電壓窗口(0 ~ 10 V),其峰值氧化電流比 25 ℃ 時(shí)的同類產(chǎn)物低 370 倍,電子電導(dǎo)率低 3 個(gè)數(shù)量級(jí)。該配位體可作為粘結(jié)劑粘結(jié)電解質(zhì)顆粒,其楊氏模量高達(dá) 208.45 GPa;該模量是同類電解質(zhì)的兩倍,可適應(yīng)鋰沉積和剝離過程中的持續(xù)應(yīng)力應(yīng)變釋放。憑借這些優(yōu)點(diǎn),該電解質(zhì)在 25 ℃ 時(shí)的臨界電流密度達(dá)到了破紀(jì)錄的 21.65 mA cm^-2^(是鋰離子固態(tài)電解質(zhì)最佳報(bào)告數(shù)據(jù)的兩倍),在10.83 mA cm^-2^下 穩(wěn)定循環(huán)6000 小時(shí),在10V下穩(wěn)定1000小時(shí)。工作溫度窗口寬達(dá) -30 ℃ 至 150 ℃。* 開發(fā)的鈷酸鋰電池在高壓下表現(xiàn)出卓越的可逆性。該研究結(jié)果為固態(tài)電解質(zhì)的氧化穩(wěn)定性和枝晶抑制指明了方向,為高壓鋰電池的發(fā)展帶來了巨大**的進(jìn)步。
 微軟雅黑; font-size: 16px; letter-spacing: 0.5px;" />
微軟雅黑; font-size: 16px; letter-spacing: 0.5px;" />基本介紹 編輯本段
在全固態(tài)電池(ASSBs)中使用固態(tài)電解質(zhì)(SEs)是一項(xiàng)非常有前途的技術(shù),由于其固有的安全性和穩(wěn)定性,可以克服液態(tài)電解質(zhì)的缺點(diǎn),為發(fā)現(xiàn)具有超高能量密度的新型電池化學(xué)物質(zhì)提供了機(jī)會(huì),如金屬鋰電池(例如,金屬鋰陽極與高電壓陰極配對(duì))。然而,大多數(shù)無機(jī) SE 都面臨著氧化穩(wěn)定性差(2 ~ 5 V 對(duì) Li^+^/Li)和枝晶晶形成的問題,室溫(RT)下的臨界電流密度(CCD)小于 11 mA cm^-2^,這極大地阻礙了它們的應(yīng)用。
因此,陰離子工程,如在 SE 中形成電負(fù)性更高的陰離子,可顯著提高 SE 的氧化穩(wěn)定性,從而在報(bào)道的最佳研究中實(shí)現(xiàn) 0 至 5 V 的大電壓窗口。然而,迄今為止還缺乏對(duì)更高電壓窗口的探索。最近,研究人員已經(jīng)證明,體 SE 中的高電子傳導(dǎo)性會(huì)加速氧化分解的動(dòng)力學(xué)過程,這是導(dǎo)致嚴(yán)重枝晶晶粒生長(zhǎng)的主要原因。許多研究都致力于通過添加電子導(dǎo)電率較低的第二相(如 SiO2、Al2O3、TiO2和 ZrO2)來解決干擾電子滲流問題。根據(jù)滲流理論,一旦填料的體積分?jǐn)?shù)超過某個(gè)點(diǎn),電子滲流就會(huì)被關(guān)閉,因此可以理解復(fù)合 SE 中電子電導(dǎo)率的下降。然而,這種策略的關(guān)鍵缺陷在于,體 SE 中的鋰離子電導(dǎo)率會(huì)同步降低(尤其是在 RT 條件下),而且由于惰性填料的高硬度和亮度特性,機(jī)械性能也會(huì)下降。
還有一些學(xué)者嘗試在 SEs 的晶界或電極界面中引入共價(jià)鍵分子和采用元素置換的方法,通過改變陽離子和陰離子形成離子/共價(jià)鍵無機(jī)化合物,這在一定程度上降低了 SEs 的電子傳導(dǎo)性。盡管如此,復(fù)合 SE 的熱穩(wěn)定性、化學(xué)穩(wěn)定性和電化學(xué)穩(wěn)定性仍會(huì)顯著下降,從而造成與鋰陽極的界面不相容,導(dǎo)致自發(fā)反應(yīng)和結(jié)構(gòu)退化。
在無機(jī) SE 中,LiBH4與金屬鋰相比具有獨(dú)特的熱力學(xué)穩(wěn)定性,可保持低分子量、易變形和可壓縮性,從而實(shí)現(xiàn)高能量密度和界面兼容性。然而,它們的氧化穩(wěn)定性較差(相對(duì)于 Li^+^ /Li 氧化電壓小于 2.0 V),并且在 125 ℃ 時(shí) CCD 低至 2.8 mA cm^-2^,因此枝晶晶威脅嚴(yán)重。
知識(shí)要點(diǎn) 編輯本段
工作采用LiBH4和聚甲基丙烯酸甲酯(PMMA)之間的原位熔化反應(yīng)(ISMR),在 SEs 顆粒表面的原位熔化反應(yīng)層中生成共價(jià)鍵配位。這種配位在熱力學(xué)上擴(kuò)展了陰離子的氧化穩(wěn)定性,在動(dòng)力學(xué)上阻斷了電子的滲流,起到了粘結(jié)劑的作用,將 SEs 粘結(jié)在一起,實(shí)現(xiàn)了優(yōu)異的機(jī)械性能。因此,在較寬的工作溫度范圍內(nèi),可以實(shí)現(xiàn)前所未有的高電壓工作穩(wěn)定性和枝晶晶粒抑制。
ISMR 改性電解質(zhì)的結(jié)構(gòu)表征 ISMR 改性 SE 是通過與 γ-Al2O3、LiI 和 x wt~.% PMMA(x = 0,5)高能球磨 96 小時(shí)合成的,標(biāo)記為 xPMMA。γ-Al2O3和LiI的引入是為了提高LiBH4~ SEs在RT下的鋰離子傳導(dǎo)性。
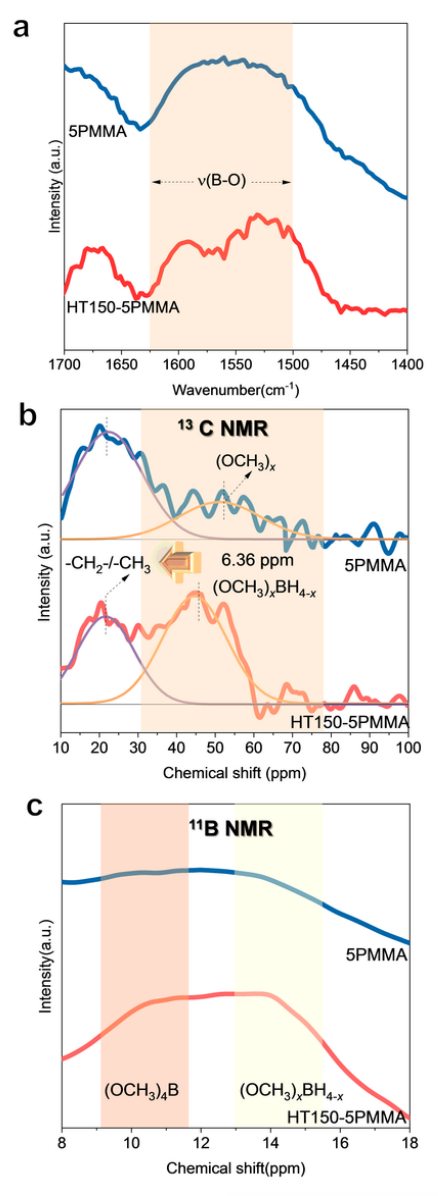
圖 1 ISMR 改性 SE 的結(jié)構(gòu)特征。
ISMR 改性電解質(zhì)的穩(wěn)定電壓窗口和枝晶抑制能力
氧化穩(wěn)定性差是 LiBH4面臨的一大挑戰(zhàn)。為了評(píng)估 HT150-0PMMA 和 HT150-5PMMA 的電壓窗口,作者采用了三次連續(xù)循環(huán)伏安 (CV) 測(cè)試,從開路電壓 (OCV) 到 10.0 V(相對(duì)于 Li^+^/Li),然后到 -0.2 V,再回到 OCV,掃描速度為 0.1 mV s^-1^。在第一個(gè)循環(huán)中,HT150-0PMMA 從 2.4 V 開始發(fā)生嚴(yán)重氧化,在 10 V 時(shí)達(dá)到 345.20 μA 的峰值電流(圖 2a)。在 HT150-0PMMA 的后續(xù)循環(huán)中,氧化過程一直持續(xù)。在 HT1500PMMA 的第 1 個(gè)循環(huán)(圖 2b 和 c)、第 2 個(gè)循環(huán)和第 3 個(gè)循環(huán)中,分別在 2.8、4.2 和 10.0 V 的截止電位下收集HT1500PMMA 的原位 B 1s 和 O 1s X 射線光電子能譜 (XPS) 譜。B1s XPS 譜(圖 2b)中的原生 BH(185.94 eV)逐漸減小,伴隨著高電壓下氧化 BH4^-^(186.63 eV)的增加,表明氧化嚴(yán)重。第 2 和第 3 個(gè)周期的氧化分解情況較差。HT150-0PMMA 的 O 1s XPS 譜在三個(gè)循環(huán)中沒有明顯變化(圖 2c)。HT1505PMMA 在前 3 個(gè)周期中的電壓窗口出乎意料地高于 HT150-0PMMA,從 - 0.2 到 10.0 V(圖 2d),氧化電流峰值為 0.93 μA,比 HT150-0PMMA 低 370 多倍。圖 2e 和 f)、第 2 個(gè)和第 3 個(gè)周期 HT150-5PMMA 在 2.8、4.2 和 10 V 電壓下的原位 B1s 和 O1s XPS 譜。圖 2e 顯示,在第 1 個(gè)循環(huán)中,氧化的 BH4^-^ 在 2.8、4.2 和 10.0 V 幾乎可以忽略不計(jì),在第 2 和第 3 個(gè)循環(huán)中也可以觀察到類似的情況,這可以通過 XPS 定量得到證實(shí)。在第 3 個(gè) CV 循環(huán)后,10.0 V 電壓下的氧化 BH4^-^為 8.91 at%,比相同測(cè)試條件下的 HT150-0PMMA 低 6 倍。更有趣的是,在第 1 個(gè)周期(圖 2f)、第 2 個(gè)周期(補(bǔ)充圖 3f)和第 3 個(gè)周期(補(bǔ)充圖 3h)的截止電壓為 2.8、4.2 和 10.0 V 時(shí),O 1s XPS 圖譜中的 (OCH3)xBH4-x幾乎保持不變,這表明 (OCH3)xBH4-x在高壓操作下相當(dāng)穩(wěn)定。
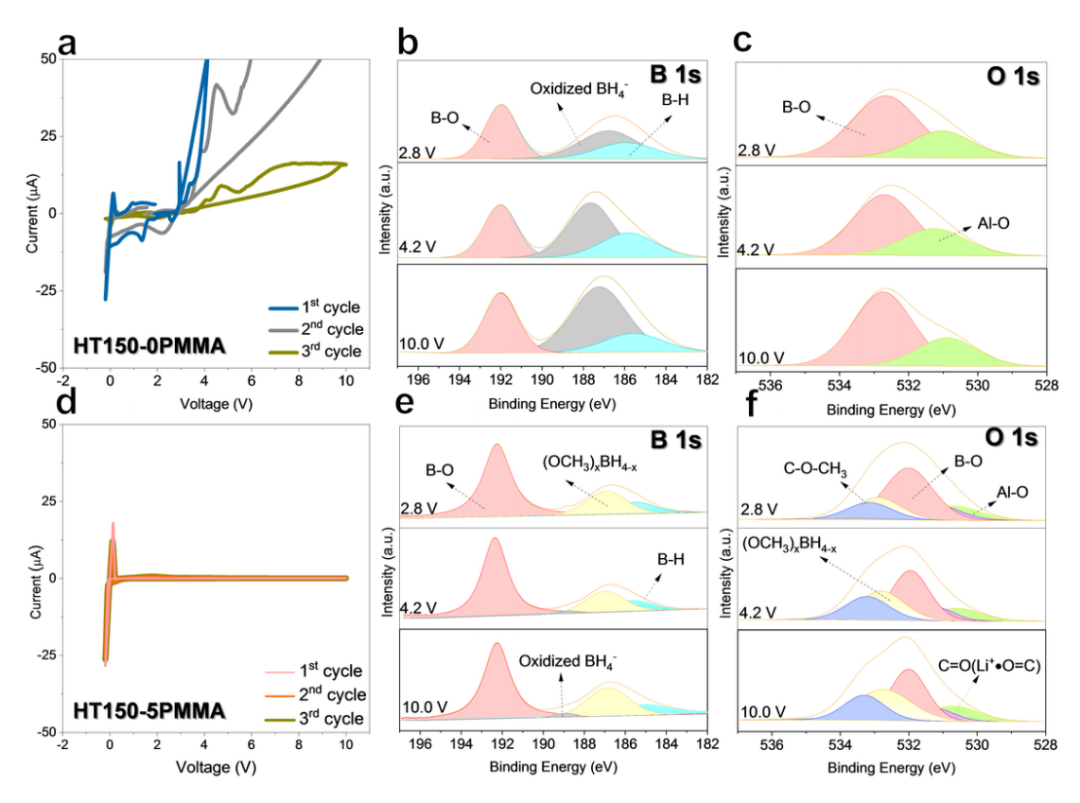
圖 2 ISMR 改性 SE 的電化學(xué)穩(wěn)定窗口。
8.0 ~ 15.0 V 的擴(kuò)展 CV 操作表明,HT150-5PMMA 的上限電壓窗口為 10.38 V,是所有基于 LiBH4的 SE 的最佳電壓窗口的兩倍。
為評(píng)估 HT150-0PMMA 和 HT150-5PMMA 的氧化穩(wěn)定性,在較寬的溫度窗口(-30 ~ 150 ℃)內(nèi)進(jìn)行了 17 次 CV 測(cè)試。HT150-0PMMA 在 -30、0、80 和 110 ℃ 出現(xiàn)不可逆氧化分解,在 130 ℃ 和 150 ℃ 直接失效。相比之下,HT150-5PMMA 在 -30、0、80、110、130 和 150 ℃ 下保持了 0 至 10.0 V 的高氧化穩(wěn)定性,陰離子和氧化分解電流可忽略不計(jì)。
在 25 ℃ 下,HT150-5PMMA 中的 CCD(21.65 mA cm^-2^)是基于 LiBH4的 SE 中第一個(gè)也是最好的 CCD,是所有鋰離子 SE 在 RT 下最佳 CCD(11 mA cm^-2^)的兩倍。
HT150-0PMMA 和 HT150-5PMMA 的 CCD 在較寬的工作溫度窗口中進(jìn)行了測(cè)試(圖 3f)。這一結(jié)果表明,HT150-5PMMA 在 -30 ℃ 至 150 ℃ 的溫度范圍內(nèi)具有優(yōu)異的枝晶抑制能力,在每個(gè)測(cè)試溫度下的 CCD 都比 HT150-0PMMA 高 10 倍,特別的在超低溫 -30 ℃(1.05 mA cm^-2^)和高溫 150 ℃(342.00 mA cm^-2^)下。HT150-5PMMA 在寬工作溫度窗口下的樹突抑制表現(xiàn)出前所未有的適應(yīng)性。
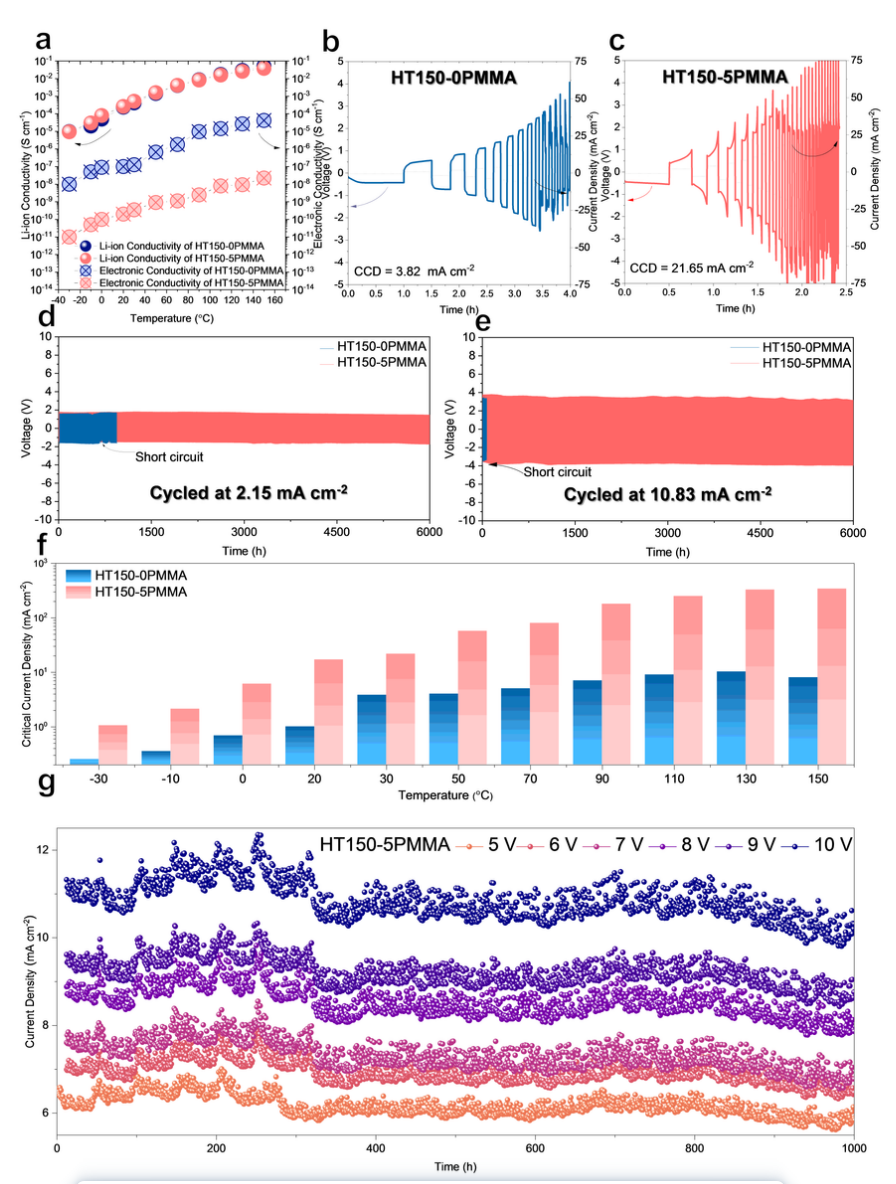
圖 3 ISMR 改性 SE 的枝晶抑制。
通過第一原理密度泛函理論(DFT)計(jì)算,以揭示 PMMA 與 LiBH4之間的相互作用。計(jì)算了 LiBH4和 PMMA 構(gòu)型的優(yōu)化結(jié)構(gòu)和分子靜電勢(shì)面映射。在圖 5a 中,電子密度聚集在 BH4 陰離子(紅色)周圍。因此,在如此高的濃度下,局部電子趨于離域。根據(jù)計(jì)算,這種構(gòu)型的第一電離能(從結(jié)構(gòu)中失去一個(gè)電子所需的能量)為 4.81 eV。相反,隨著 H^0^ 損失的增加,BHx^-^中的 B 可以牢固地與 -OCH3中的 O 配位(圖 5b-e),從而導(dǎo)致整個(gè)構(gòu)型中主要分布在 BH4^-^上的局部電子密度重新分布。因此,失去 1、2、3 和 4 個(gè) H^0^后,LiBH4和 PMMA 的第一次電離能分別為 5.73、6.52、7.09 和 9.82 eV,這表明電子泄漏越來越難以從 BHx^-^ 中產(chǎn)生,從而表現(xiàn)出很強(qiáng)的電子局域性。
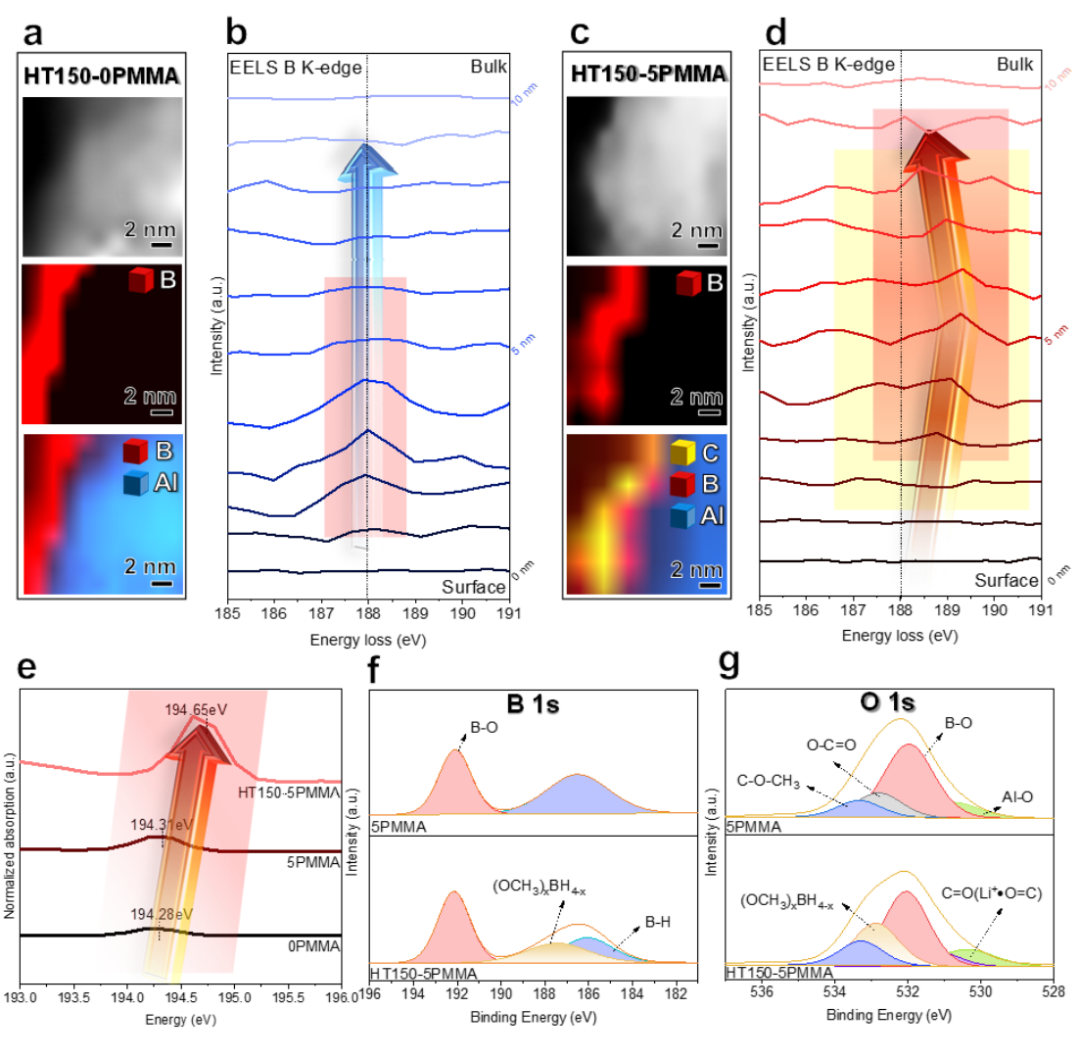
圖 4 ISMR 改性 SE 中 (OCH3)xBH4-x的形成機(jī)制。
為了研究 (OCH3)xBH4-x在提高電化學(xué)性能特性方面的作用,根據(jù) 100 至 150 ℃ 的不同反應(yīng)溫度合成了一系列 HTy-5PMMA,以獲得不同原子比的 (OCH3)xBH4-x。隨后,測(cè)量了 (OCH3)xBH4-x、初始氧化電壓、電子電導(dǎo)率和 CCD 之間的關(guān)系,如圖 5f 所示。顯然,隨著 (OCH3)xBH4-x的增加,SEs 的初始氧化電壓呈現(xiàn)出從 1.80 V 到 10.38 V 的線性增長(zhǎng),這證實(shí)了由于 (OCH3)xBH4-x在 SEs 中的強(qiáng)電子局域化,BH4^-^氧化過程中的電子交換可以在熱力學(xué)上被關(guān)閉,這與 DFT 計(jì)算結(jié)果一致。此外,在圖 5f 中,隨著 (OCH3)xBH4-x的增加,電子傳導(dǎo)性降低了幾個(gè)數(shù)量級(jí),這證實(shí)了由于 (OCH3)xBH4-x在塊狀 SEs 粒子表面的生成量增加,電子穿透可以被有效阻斷。因此,CCD 呈線性增長(zhǎng),與電子導(dǎo)電率下降的趨勢(shì)相反。因此,作者可以得出結(jié)論:氧化穩(wěn)定性的增強(qiáng)和枝晶的抑制可歸因于 ISMR 原位生成的 (OCH3)xBH4-x。
HT1505PMMA 在循環(huán)前以及循環(huán) 500 小時(shí)和 5000 小時(shí)后均未出現(xiàn)明顯的裂紋、缺陷或鋰沉積,這證實(shí)了其在長(zhǎng)期鍍鋰和剝離過程中具有優(yōu)異的結(jié)構(gòu)完整性。
(OCH3)xBH4-x可作為 LiBH4SEs 顆粒表面的粘結(jié)劑,將 SEs 粘結(jié)成更緊湊的結(jié)構(gòu),從而大大增強(qiáng)了塊體 SEs 的彈性行為,以適應(yīng)循環(huán)過程中持續(xù)的應(yīng)變應(yīng)力釋放。
通用研究 編輯本段
為了研究作者的發(fā)現(xiàn)的通用性,作者進(jìn)一步測(cè)量了 Li2B12H12和 HT150-Li2B12H12-5PMMA 的電化學(xué)性能特征,它們是通過與 HT150-5PMMA 相同的反應(yīng)合成的。結(jié)果表明,HT150-Li2B12H12-5PMMA在25 ℃時(shí)的CCD高達(dá)4.78 mA cm^-2^,是原生Li2B12H12的約6倍。在 LiAlH4和 LiNH2體系中也可以發(fā)現(xiàn)類似的趨勢(shì);不過,CCD 只增加了三倍。此外,所有經(jīng) ISMR 修飾的氫化物 SE 都能提供接近 10 V 的高度穩(wěn)定的電壓窗口,氧化電流相對(duì)于其原始形態(tài)幾乎可以忽略不計(jì)。
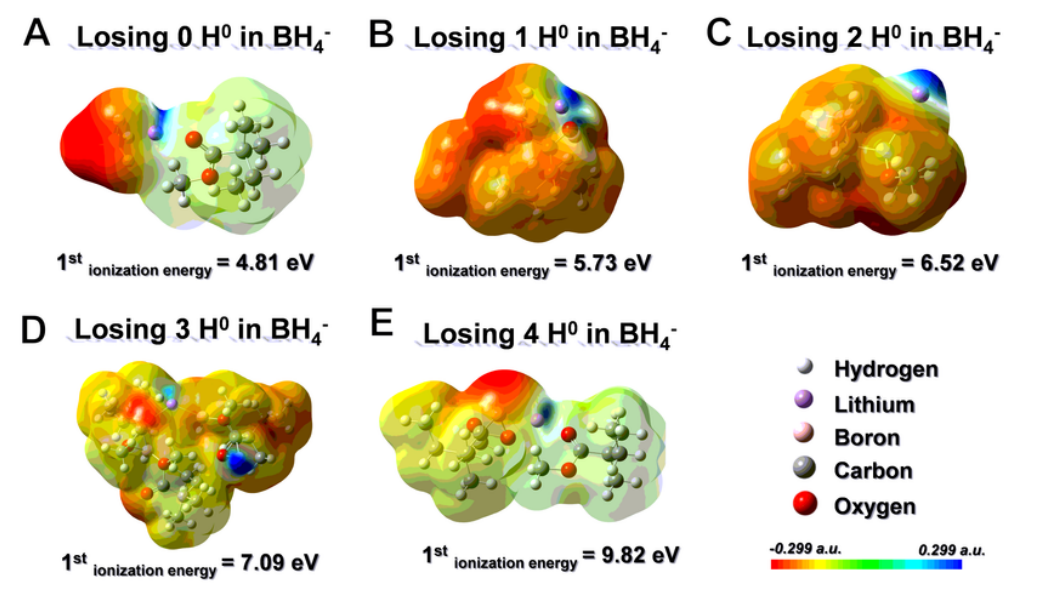
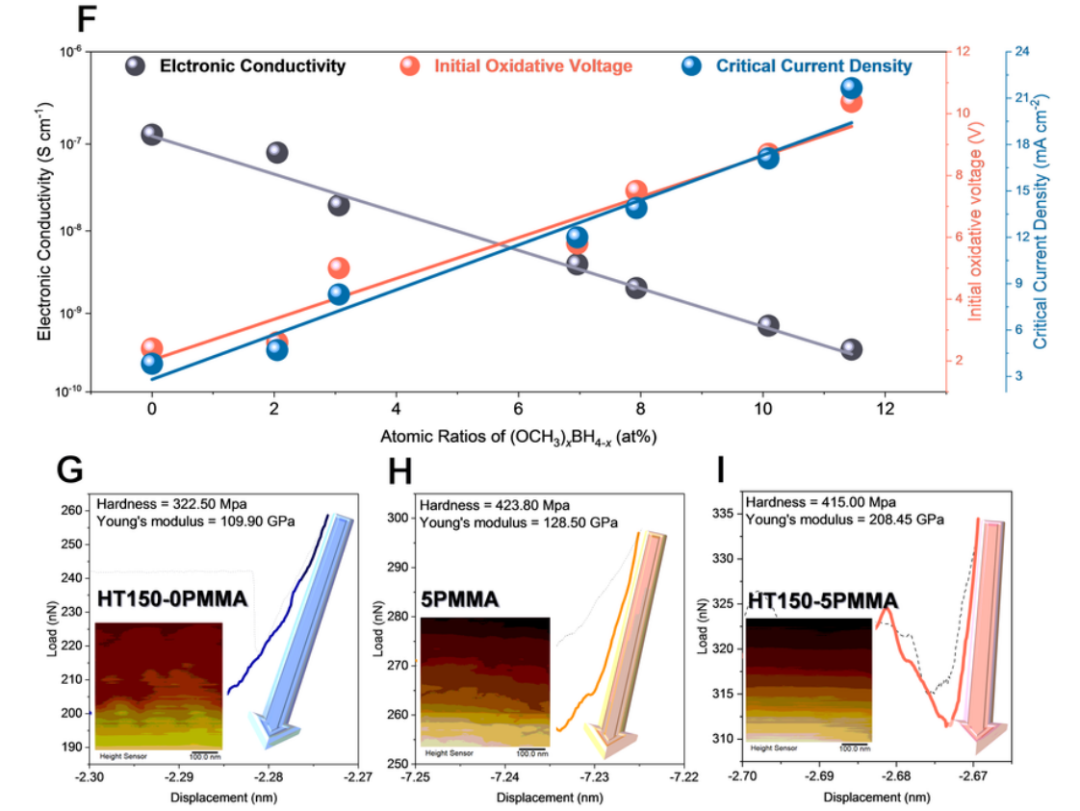
圖 5 (OCH3)xBH4-x在提高電化學(xué)性能方面的作用。
使用 ISMR 改性電解質(zhì)的高壓鋰金屬電池
使用鋰和鈷酸鋰作為電極,在 25 ℃ 下組裝并測(cè)試了高壓 ASSB。為了避免 LiBH4和 LiCoO2之間不可避免的反應(yīng),利用濕化學(xué)方法在 LiCoO2表面涂覆了 2 wt~.% 的 Li3InCl6~。使用 HT150-5PMMA 合成的 ASSB 初始比容量為 114.6 mAh g^-1^,在 0.1 C 下循環(huán) 100 次后的最高保持率為 95.8%(圖 6a 和 b)。相比之下,使用 HT150-0PMMA 合成的 ASSB 的初始放電比容量較低,為 72.3 mAh g^-1^,隨后容量迅速衰減,最終在 45 個(gè)循環(huán)后失效。
令人驚訝的是,含有 HT150-5PMMA 的鈷酸鋰 ASSB 實(shí)現(xiàn)了 111.4 mAh g^-1^ 的高初始放電容量,在工作電壓為 3.0 至 4.2 V 的條件下,循環(huán) 200 次后的容量保持率為 100%,在 0.5 C 條件下循環(huán) 300 次后的容量保持率為 94.2%。
涉及 HT150-5PMMA 的高壓鈷酸鋰 ASSB 在 3.0 至 4.2、4.6、4.8、5.0、8.0 和 10.0 V 的電壓窗口中進(jìn)一步循環(huán)。充放電曲線和相應(yīng)的循環(huán)性能特征分別如圖 6e 和 f 所示。作者的 HT150-5PMMA 在 3.0 至 4.6、4.8 和 5.0 V 的電壓范圍內(nèi)具有較高的可逆性,在 25 ℃(0.1 C)條件下的可逆初始放電容量分別為 128.7、135.0 和 144.3 mAh g^-1^。然而,由于鈷酸鋰在高 SOC 下過度脫鋰導(dǎo)致結(jié)構(gòu)不可逆崩潰,45 個(gè) ASSB 循環(huán)一次,在 8 V 和 10 V 的上限截止電壓下分別顯示 130.5 和 136.0 mAh g^-1^ 的初始放電容量,這表明 ISMR 改性 SE 在高壓鋰金屬 ASSB 中大有可為。
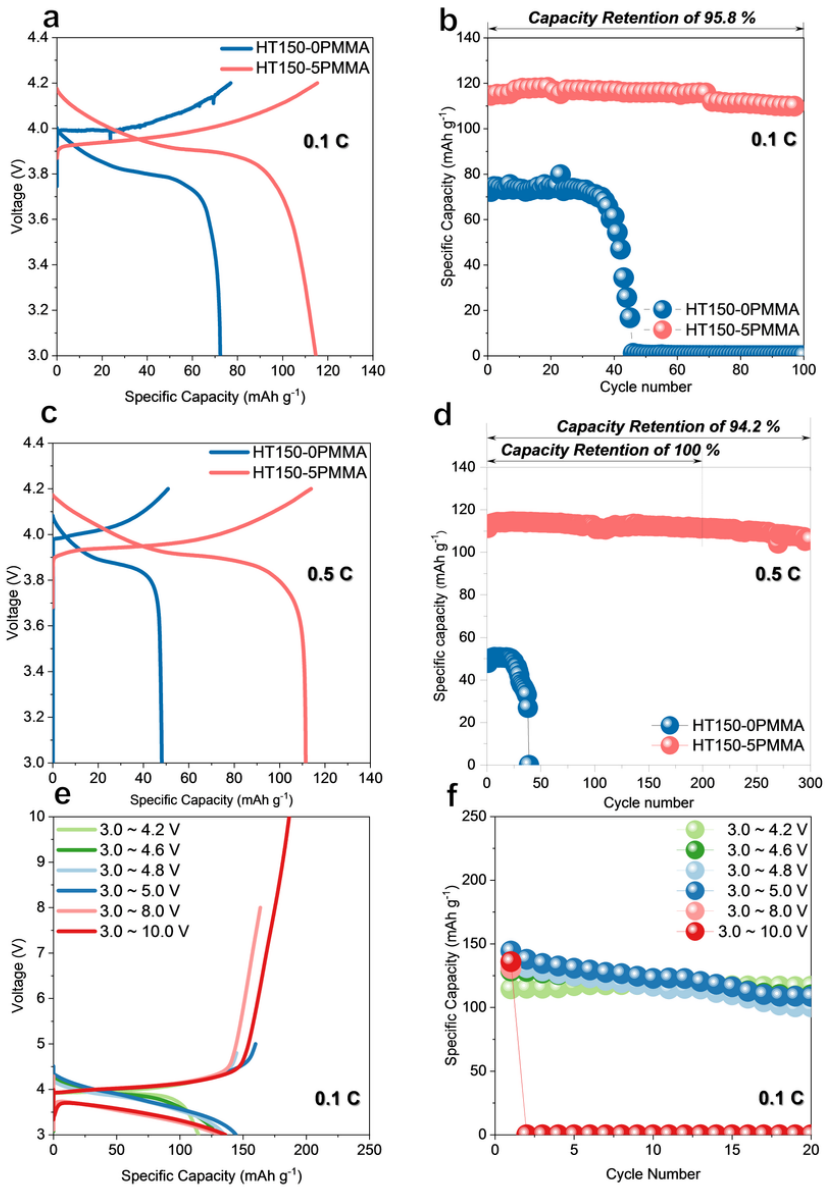
圖6 使用 ISMR 改性 SE 構(gòu)建的高電壓Li-LCO ASSB。
反應(yīng)策略 編輯本段
工作開發(fā)一種新穎的原位熔化反應(yīng)策略,在 LiBH4SEs 的顆粒表面生成共價(jià)鍵配位,以解決 SEs 氧化穩(wěn)定性差和枝晶問題。這種配位從熱力學(xué)角度提高了陰離子的內(nèi)在氧化穩(wěn)定性,并從動(dòng)力學(xué)角度阻止了 SEs 粒子表面的電子傳導(dǎo),從而抑制了枝晶的生長(zhǎng)。此外,它還起到粘結(jié)劑的作用,有助于在鋰的連續(xù)沉積和剝離過程中實(shí)現(xiàn)出色的機(jī)械性能,以適應(yīng)應(yīng)力應(yīng)變的釋放。因此,所獲得的 SE 保持了創(chuàng)紀(jì)錄的高電壓窗口(0 ~ 10 V),峰值氧化電流比同類產(chǎn)物低 370 倍;此外,在 25 ℃ 下具有前所未有的 CCD(21.65 mA cm^-2^),在 10.83 mA cm^-2^ 下具有 6000 小時(shí)的超長(zhǎng)循環(huán)穩(wěn)定性,在 10 V、25 ℃ 下具有 1000 小時(shí)的超長(zhǎng)循環(huán)穩(wěn)定性,實(shí)現(xiàn)了從 - 30 ℃ 到 150 ℃ 的寬工作溫度窗口。他們的鈷酸鋰電池在 3.0 至 4.2 V、60 mA g^-1^ 的條件下循環(huán) 200 次后,容量保持率達(dá)到 100%,并且在 3.0 至 5.0 V 范圍內(nèi)具有可逆循環(huán)穩(wěn)定性。作者的發(fā)現(xiàn)為促進(jìn)高能量密度 ASSB SE 的電化學(xué)穩(wěn)定性提供了一個(gè)清晰的視角。
附件列表
詞條內(nèi)容僅供參考,如果您需要解決具體問題
(尤其在法律、醫(yī)學(xué)等領(lǐng)域),建議您咨詢相關(guān)領(lǐng)域?qū)I(yè)人士。